Add a Pipeline with parallel computing support#
In this chapter we’ll add a second Pipeline to our plugin, and then we’ll add parallel computing support to that Pipeline.
This will enable your users to utilize multi-processor computers or multi-node high performance computing infrastructure (e.g., computer clusters) to analyze their data.
tl;dr
The complete code that I wrote to add the second
Pipelineto my plugin can be found here: caporaso-lab/q2-dwq2.The code that I developed to add parallel computing support to the new
Pipelinecan be found here: caporaso-lab/q2-dwq2.Finally, I added a usage example that can be used to compare the performance of serial and parallel runs of the new Pipeline here: caporaso-lab/q2-dwq2. Because the third commit includes (a small amount) of real data, as opposed to the toy-sized data we’ve been using in unit tests, it increases the runtime of the test suite considerably (from about 30 seconds to about 6 minutes, on the M3 MacBook Pro that that I’m developing on).
Add a local alignment search Pipeline#
The first goal of this section is to define a new Pipeline that performs a local alignment search and then tabulates those results for the user to view.
This is effectively the same goal as the ubiquitous bioinformatics tool, BLAST [5].
The underlying theory, and the implementation that we’ll use here, are presented in the Sequence Homology Searching chapter of An Introduction to Applied Bioinformatics [3].
Briefly, given one or more query sequences and one or more reference sequences, a local alignment search uses pairwise sequence alignment to identify the most similar reference sequence for each query sequence. In our implementation, as in the BLAST implementation, we’ll allow a query sequence to match a subsequence of a reference sequence (this is useful, for example, if the query sequence represents a fragment of gene, and the reference sequences represent full-length genes). To achieve this, the underlying alignment algorithm that we’ll use is Smith-Waterman local pairwise alignment [6], which is presented in the Pairwise Sequence Alignment chapter of An Introduction to Applied Bioinformatics.
The output for a typical local alignment search is a table of the results.
Depending on what the user requests, this can describe simply the best match in the database for each query (often defined as the reference sequence which obtains the highest scoring pairwise alignment with the query sequence), or a reverse sorted list of the matches, such that for each query sequence the n best matches are presented in order from best to worst.
Depending on the implementation, some additional information may be provided about each alignment including the percent similarity between the aligned query and reference sequence, the length of the alignment, and the score of the alignment.
To add a local alignment search Pipeline to our plugin, we’re going to add one new Method and one new Visualizer.
The method will be called local_alignment_search, and it will take one or more query sequences and one or more reference sequences as inputs, and it will accept several parameters used to control the behavior of the alignment algorithm and the result tabulation.
As an output, it will generate a tabulation of the search results, grouped by query id and sorted in descending order of alignment score.
This table will be an Artifact of a new class, LocalAlignmentSearchResults, such that these results could be used by another action (e.g., one that associates metadata with the query sequences, such as taxonomic origin or gene function, based on metadata associated with their best matching reference sequences).
This means that we’ll also create a new format, some new transformers, and a new artifact class in support of the new functionality.
The new visualizer, tabulate_las_results (where las is an abbreviation of local alignment search) will take a LocalAlignmentSearchResults artifact as input and will produce a user-friendly view of it.
Finally, the new pipeline, search_and_summarize, will link local_alignment_search and tabulate_las_results together, returning both the LocalAlignmentSearchResults data artifact and the human-readable visualization.
Since you’ve already done all of this type of work before for other functionality in your plugin, we won’t go through this in detail. Use this as an opportunity to read and interpret QIIME 2 plugin code. Review the code added in caporaso-lab/q2-dwq2 and add it to your plugin.
Add parallel computing support to search_and_summarize#
QIIME 2’s formal support for parallel computing makes use of Parsl, and enables developers to create parallel Pipelines that can run on compute resources ranging from multi-core laptops to multi-node high performance computer clusters.
Parallel Pipelines follow the split-apply-combine strategy.
In the split stage, input data is divided into smaller data sets, generally of the same type as the input.
Then, in the apply stage, the intended operation of the Pipeline is applied to each of the smaller data sets in parallel.
Finally, in the combine stage, the results of each apply operation are integrated to yield the final result of the operation as a single data set, generally of the same type as the output of each apply operation.
In the context of search_and_summarize, this can work as follows (see the flow chart for a visual summary).
In the split stage, the query sequences can be divided into roughly equal sized splits of sequences, such that each query sequence appears in only one split.
In the apply stage, the reference sequences and each split of query sequences can be provided as the input in parallel calls to the local_alignment_search method.
Each parallel call will result in a tabular output of search results for its split of the query sequences against the full set of reference sequences.
This has the effect of reducing the number of query sequences that are searched against the reference in a given call to local_alignment_search.
Finally, in the combine stage, each of the tabular outputs are joined, and the resulting table is sorted and filtered to the top n hits per query.
This enables the slow step in this workflow - local_alignment_search - to be run in parallel on different processors.
Parsl takes care of all of the work of sending each apply job out to different processors and monitoring them to see when they’re all done.
Our work on the plugin development side, after we’ve already defined the apply operation (local_alignment_search, in this example), is to define the actions that will perform the split and combine operations.
These operations will be new QIIME 2 Methods.
split-apply-combine flowchart for search and summarize
flowchart TD
A["query: FeatureData[Sequence]"] ----> B{"split-sequences"}
B -- split 0 --> C["FeatureData[Sequence]"]
B -- split 1 --> D["FeatureData[Sequence]"]
B -- split 2 --> E["FeatureData[Sequence]"]
B -- split 3 --> F["FeatureData[Sequence]"]
subgraph "Collection[FeatureData[Sequence]]"
C
D
E
F
end
C ----> G{"local-alignment-search\n(apply step)"}
D ----> H{"local-alignment-search\n(apply step)"}
E ----> I{"local-alignment-search\n(apply step)"}
F ----> J{"local-alignment-search\n(apply step)"}
G ----> K["LocalAlignmentSearchResults"]
H ----> L["LocalAlignmentSearchResults"]
I ----> M["LocalAlignmentSearchResults"]
J ----> N["LocalAlignmentSearchResults"]
subgraph "Collection[LocalAlignmentSearchResults]"
K
L
M
N
end
K ----> O{"combine-las-results"}
L ----> O
M ----> O
N ----> O
O ----> P["LocalAlignmentSearchResults"]
P ----> Q{"tabulate-las-results"}
Q ----> R((("Visualization")))
S["reference: FeatureData[Sequence]"] -.-> G
S -.-> H
S -.-> I
S -.-> J
subgraph Key
T["Artifact"]
U{"Action"}
V((("Visualization")))
end
Defining a split Method#
The split, apply, and combine actions are all QIIME 2 Methods, like any others. Our split method will build on a function that takes sequences as input, along with a variable defining the number of sequences that should go in each split, and it will result a dictionary mapping an arbitrary split identifier to a split of sequences. This can look like the following:
# Store the default values for `split_sequences` in a dict, so we can
# reference them in multiple places.
_split_seqs_defaults = {
'split_size': 5
}
def split_sequences(
seqs: DNAIterator,
split_size: int = _split_seqs_defaults['split_size']) \
-> DNAIterator:
result = {i : DNAIterator(split)
for i, split in enumerate(_batched(seqs, split_size))}
return result
Here, the input DNAIterator is passed to a function, _batched, which yields subsets of the input iterator each with split_size sequences.
(If the number of input sequences isn’t a multiple of split_size, the last iterator in result will have fewer than split_size sequences.)
The _batched function does most of the work of splitting up the sequences here.
Referring to the _methods.py file in caporaso-lab/q2-dwq2, add the split_sequences and _batched functions to your _methods.py file.
This is also a good time to write unit tests for your split_sequences function.
I recommend designing and implementing the unit tests yourself, but you can also refer to the tests that I wrote in test_methods.py.
Should our sequence splitter take the size of each split or the number of splits to create as input?
Defining how the splits should be created is a design decision that the developer must consider.
If having the user provide the number of sequences that should go into each split (as we are here), they will need to know the length of seqs to make the best decision for the computer where they will run the job.
For example, if they have 16 processors available to them, it might make the most sense to have around 15 splits, so they’ll need to know the number of sequences they’re providing as input and divide that by 15 (rounding fractional values up to the next whole number).
If instead, we have the user define how many splits they want (e.g., by providing a num_splits value), the computer will need to know how many sequences to put in each split, so it will need to know the length of seqs to make a good decision.
That will require either taking two passes through seqs - first to find out how many sequences there are, and second to do the actual splitting - or reading the entirety of seqs into memory (e.g., calling list(seqs)).
The first of these two options can be slow if seqs is long, and the second can be problematic if the size of seqs is larger than the amount of available RAM.
The best option may be to let the user choose between these two approaches by either providing split_size or num_splits. If providing num_splits will be problematic due to runtime or required memory, they can opt to figure out the split_size themselves and provide it.
For the sake of this example I’m going with the simplest option of just requiring the user to provide the split_size.
As an exercise after completing this section, implement the alternative approach of allowing the user to either provide the split_size or num_splits.
Registering split_sequences#
split_sequences will be used in our search_and_summarize Pipeline, and we therefore need to register it as a new Action on our plugin.
This is done in the typical way, and at this point you’re probably getting fairly comfortable with this.
The one thing that is needed here, but which we haven’t encountered yet in other Actions, is allowing for an arbitrary number of outputs: the individual splits of sequences.
The combination of the number of sequences in an input and the user-specified split_size will define how many outputs will be generated by a call to this action, so it’s not possible for us to determine this when registering the action.
We therefore define our output type as a Collection of FeatureData[Sequence] artifacts, which we annotate as Collection[FeatureData[Sequence]] in the outputs dictionary.
To achieve this, import Collections from qiime2.plugin at the top of your plugin_setup.py file.
In your call to plugin.methods.register_function, you can then pass:
outputs={'splits': Collection[FeatureData[Sequence]]}
Using that tip to handle the generation of an arbitrary number of outputs, refer to the other actions that you’ve registered, and start implementing the registration step without referring to the code in q2-dwq2.
Once you’ve done that and have it working, take a look at my call to plugin.methods.register_function in the plugin_setup.py file of q2-dwq2 to see if you missed anything that I included (I added the code in this commit: caporaso-lab/q2-dwq2).
Defining and registering a combine method#
Our combine method is effectively performing the opposite operation relative to our split method.
In this case, we need a method that takes multiple LocalAlignmentSearchResults Artifacts as input, and returns them in a single, combined LocalAlignmentSearchResults Artifact.
Let’s create a new combine_las_reports function.
def combine_las_reports(reports: pd.DataFrame) -> pd.DataFrame:
results = pd.concat(reports.values())
return results
In this function, reports, which is a dict of one or more pandas DataFrames, are combined using the pandas.concat function, which concatenates DataFrames in the order in which they’re received.
The resulting object is a single DataFrame, which we’ll return from this function.
Note
Something you might notice when looking at this function’s signature is that the view type for the collection of local alignment search reports (the reports variable) is pd.DataFrame, not a collection of DataFrames as you might expect.
QIIME 2 doesn’t differentiate between single inputs and collections of inputs in the type annotations of functions to be registered as Actions.
This was also apparent above, when split_sequences was annotated as returning a DNAIterator, which will always be a Python dictionary where DNAIterator objects are the values.
This is just something to be aware of.
The last remaining things to do before we parallelize search-and-summarize is to write unit tests of combine_las_reports, and register a combine-las-reports Method in the plugin.
Complete these steps as an exercise now, and then refer to my code (caporaso-lab/q2-dwq2) to check your work.
One thing that I hope is evident from this section is that split and combine functions tend to be pretty simple. These operations can be more complex in some cases (at some point, I’ll add an example of that), but often they’re very simple.
Update search-and-summarize to use split and combine Methods#
We’re now ready to update search-and-summarize to run in parallel.
First, we’ll add the split_size parameter to search_and_summarize, so we can pass it through to split_sequences.
def search_and_summarize(
...
split_size=_split_seqs_defaults['split_size'],
...
Then, inside the function, we’ll get the new Actions we just defined:
...
split_action = ctx.get_action('dwq2', 'split_sequences')
combine_action = ctx.get_action('dwq2', 'combine_las_reports')
...
Then, we’ll apply the split action to our input sequences:
query_splits, = split_action(query_seqs, split_size=split_size)
And next, the interesting bit happens.
We start by defining a list (las_results) to collect our local alignment search results for each split of the query sequences.
Then we iterate over the splits, applying the local alignment search method to each split, and appending the results in the las_results list that we just created.
las_results = []
for q in query_splits.values():
las_result, = las_action(
q, reference_seqs, n=n, gap_open_penalty=gap_open_penalty,
gap_extend_penalty=gap_extend_penalty, match_score=match_score,
mismatch_score=mismatch_score)
las_results.append(las_result)
Finally we combine all of the individual results, and from this point everything proceeds as it did before we added parallel support.
las_results, = combine_action(las_results)
Under the hood is where the magic happens here.
Because we’re iterating over a collection of QIIME 2 Artifacts (the for loop above) in a QIIME 2 Pipeline, each las_result object is a proxy for a real QIIME 2 artifact.
The jobs to create them are distributed as indicated in the user’s QIIME 2 parallel configuration.
The code continues executing as if these were real Artifacts, not proxy Artifacts, until something is needed from them.
At that point, the code will block (i.e., wait) until the proxy Artifacts become real Artifacts, and then continue processing.
Pipeline resumption ♻️
A cool feature that you get for free here is pipeline resumption.
If your Pipeline is interrupted mid-run (for example, because the jobs ran out of alloted time or memory on the shared compute cluster they’re running on), users can restart the job and all Results that were already computed will be recycled and not need to be computed again.
This can save your users a lot of time and frustration, and reduce unnecessary utilization of compute resources and the energy used to power that computation.
The last steps before this is ready to use are to update the parameters to search_and_summarize in plugin_setup.py, and to add a test of the parallel execution of the Pipeline. Make the necessary changes to plugin_setup.py, and then we’ll add a new unit test.
Testing the parallel Pipeline#
To test that the parallel functionality works as expected, we’ll make some adaptations to our test of the Pipeline. Specifically, we’ll reuse most of our test of the serial functionality and confirm that when the tests are run in parallel they still work as expected.
There are a lot of changes in the test_pipelines.py file in the commit associated with this section, but most of them are just refactoring the code to reuse the input Artifacts and expected test results.
First, I added a setUp method to the SearchAndSummarizeTests.
setUp is a special method that runs before each individual test function, so it’s useful for sharing information across tests.
In this new function, the first thing I do is call super().setUp(), which calls the setUp function on the base class (TestPluginBase in this case), if one exists.
This ensures that any upstream configuration is still happening.
Then, I moved the code from test_simple1 that accessed the Pipeline and created the input Artifacts to this function and set all of these as attributes on the SearchAndSummarizeTests class.
Now I can access these via the self variable in the methods of this class.
After all of these changes, my class definition starts as follows:
class SearchAndSummarizeTests(TestPluginBase):
package = 'q2_dwq2.tests'
def setUp(self):
super().setUp()
self.search_and_summarize_pipeline = \
self.plugin.pipelines['search_and_summarize']
query_sequences = [skbio.DNA('ACACTCTCCACCCATTTGCT',
metadata={'id': 'q1'}),
skbio.DNA('ACACTCACCACCCAATTGCT',
metadata={'id': 'q2'})]
query_sequences = DNAIterator(query_sequences)
self.query_sequences_art = qiime2.Artifact.import_data(
"FeatureData[Sequence]", query_sequences, view_type=DNAIterator
)
reference_sequences = [
skbio.DNA('ACACTCACCACCCAATTGCT', metadata={'id': 'r1'}), # == q2
skbio.DNA('ACACTCTCCACCCATTTGCT', metadata={'id': 'r2'}), # == q1
skbio.DNA('ACACTCTCCAGCCATTTGCT', metadata={'id': 'r3'}),
]
reference_sequences = DNAIterator(reference_sequences)
self.reference_sequences_art = qiime2.Artifact.import_data(
"FeatureData[Sequence]", reference_sequences, view_type=DNAIterator
)
The next thing I did was define a helper function that compares the observed results to the expected results.
Like the creation of the Artifacts above, this code was also moved from test_simple1.
def _test_simple1_helper(self, observed_hits, observed_viz):
expected_hits = pd.DataFrame([
['q1', 'r2', 100., 20, 40., 'ACACTCTCCACCCATTTGCT',
'ACACTCTCCACCCATTTGCT'],
['q1', 'r3', 95., 20, 35., 'ACACTCTCCACCCATTTGCT',
'ACACTCTCCAGCCATTTGCT'],
['q1', 'r1', 90., 20, 30., 'ACACTCTCCACCCATTTGCT',
'ACACTCACCACCCAATTGCT'],
['q2', 'r1', 100., 20, 40., 'ACACTCACCACCCAATTGCT',
'ACACTCACCACCCAATTGCT'],
['q2', 'r2', 90., 20, 30., 'ACACTCACCACCCAATTGCT',
'ACACTCTCCACCCATTTGCT'],
['q2', 'r3', 85., 20, 25., 'ACACTCACCACCCAATTGCT',
'ACACTCTCCAGCCATTTGCT']],
columns=['query id', 'reference id', 'percent similarity',
'alignment length', 'score', 'aligned query',
'aligned reference'])
expected_hits.set_index(['query id', 'reference id'], inplace=True)
pdt.assert_frame_equal(observed_hits.view(pd.DataFrame), expected_hits)
# observed_viz is a qiime2.Visualization.
# access its index.html file for testing.
index_fp = observed_viz.get_index_paths(relative=False)['html']
with open(index_fp, 'r') as fh:
observed_index = fh.read()
self.assertIn('q1', observed_index)
self.assertIn('q2', observed_index)
self.assertIn('r1', observed_index)
self.assertIn('r2', observed_index)
self.assertIn('ACACTCACCACCCAATTGCT', observed_index)
self.assertIn('ACACTCTCCACCCATTTGCT', observed_index)
Then, I modified the name of test_simple1 to test_simple1_serial, and adapted it to use the pipeline and artifact attributes, and to call _test_simple1_helper to compare the observed and expected results.
def test_simple1_serial(self):
observed_hits, observed_viz = self.search_and_summarize_pipeline(
self.query_sequences_art, self.reference_sequences_art)
self._test_simple1_helper(observed_hits, observed_viz)
Finally, I defined a new test function test_simple1_parallel, which calls the same Pipeline with the same inputs, and compares the observed output to the expected output in the same way as test_simple1_serial.
The only difference is that in this test search_and_summarize is running in parallel instead of serially.
This is achieved by using the with ParallelConfig() context manager, and calling the Pipeline using its .parallel method.
Add the following import to the top of your test_pipeline.py file:
from qiime2.sdk.parallel_config import ParallelConfig
Then add the following test function:
def test_simple1_parallel(self):
with ParallelConfig():
observed_hits, observed_viz = \
self.search_and_summarize_pipeline.parallel(
self.query_sequences_art, self.reference_sequences_art)
self._test_simple1_helper(observed_hits, observed_viz)
After making all of the changes described here to your _pipelines.py and test_pipelines.py files, run the test suite with make test.
If all tests pass, you should be good to go.
If not, compare your code to mine (caporaso-lab/q2-dwq2) to see what’s different.
Compare the serial versus parallel run times of the search-and-summarize#
Now that we’ve added parallel computing support to our search-and-summarize Pipeline, we can try it out to see how it impacts run time.
To do this, we need a data set that is big enough that the benefits of parallelization outweigh the overhead associated with parallelization.
For example, the splitting of the query sequences and the combining of the search result tables each take some time - if our input data is tiny, that time might be longer than the time to just compute the results serially, so running the code in parallel wouldn’t result in a noticeable decrease in runtime (the parallel runtime might even be larger than the serial runtime).
In a third commit associated with this section (caporaso-lab/q2-dwq2), I added a usage example to our new Pipeline that uses a larger data set. Note that integrating this usage example does considerably increase the runtime of the unit tests. I suggest trying it out as-is, but after experimenting with it you can feel free to reduce the number of query (to 2, for example) and reference sequences (to 3, for example) to reduce the runtime, or just keep it as-is - it’s up to you.
Refer to this commit to integrate the usage example into your code.
Then, run the usage example as is to get a feel for its serial run time.
After that, call the usage example again providing the --parallel parameter to get a feel for its parallel run time.
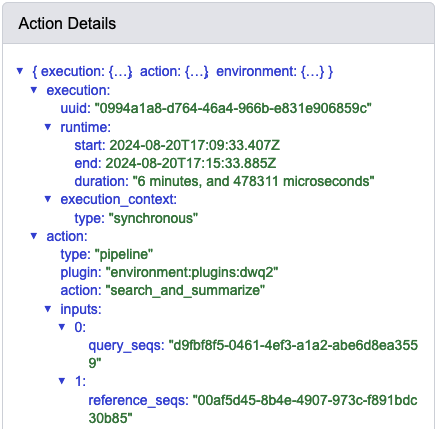
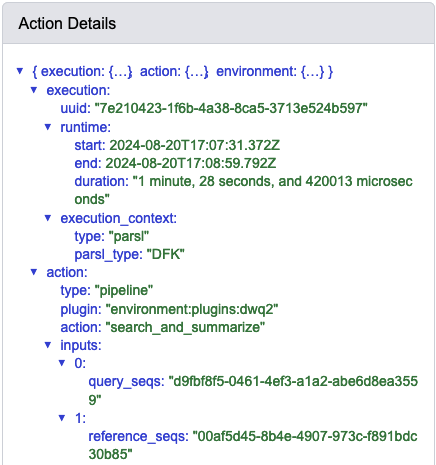
When I do this, I observe the following run times associated with the data provenance of this step: